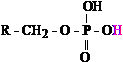Exercise-Induced
Metabolic Acidosis: Robert
A Robergs Exercise
Science Program, University of New Mexico, Albuquerque, NM 87059, USA. Email:
rrobergs@unm.edu Sportscience
5(2), sportsci.org/jour/0102/rar.htm, 2001 (7843 words) Reviewed by Lawrence Spriet,
Department of Human Biology and Nutritional Science, University of Guelph,
Ontario, Canada The
widespread belief that intense exercise causes the production of “lactic
acid” that contributes to acidosis is erroneous. In the breakdown of a
glucose molecule to 2 pyruvate molecules, three reactions release a total of
four protons, and one reaction consumes two protons. The conversion of 2 pyruvate to 2 lactate
by lactate dehydrogenase (LDH) also consumes two protons. Thus lactate
production retards rather than contributes to acidosis. Proton release also occurs during ATP
hydrolysis. In the transition to a higher exercise intensity, the rate of ATP
hydrolysis is not matched by the transport of protons, inorganic phosphate
and ADP into the mitochondria.
Consequently, there is an increasing dependence on ATP supplied by
glycolysis. Under these conditions, there is a greater rate of cytosolic proton release from glycolysis and ATP
hydrolysis, the cell buffering capacity is eventually exceeded, and acidosis
develops. Lactate production increases
due to the favorable bioenergetics for the LDH reaction. Lactate production is therefore a
consequence rather than a cause of cellular conditions that cause
acidosis. Researchers, clinicians, and
sports coaches need to recognize the true causes of acidosis so that more
valid approaches can be developed to diminish the detrimental effects of
acidosis on their subject/patient/client populations. Reprint pdf · Reprint doc KEYWORDS:
lactate, lactic acid, glycolysis, ATP, hydrolysis. |
|
The Biochemistry of Metabolic Acidosis Fundamentals of Acid-Base Physiology The Source of Protons During Catabolism In
Skeletal Muscle Phosphagen Energy System: Creatine Kinase
Reaction Phosphagen Energy System: Adenylate Kinase
Reaction Phosphagen Energy System: AMP Deaminase
Reaction Phosphagen Energy System: ATP Hydrolysis Phosphagen Energy System: Summary The Lactate Dehydrogenase Reaction The Balance of Proton Production and
Consumption in Muscle Contraction Summary of Cytosolic Proton Exchange Application of Biochemistry of Acidosis to
Exercise Physiology Why is Lactic Acid Still Thought to Cause
Acidosis? The
scientific method involves stringent criteria for the evaluation of
knowledge, but the method is not perfect.
Research findings and their interpretations can be raised prematurely
to the status of a fact. Some of these
“facts” can even become a pivotal component of a knowledge base, termed a
construct. Consequently, continual
re-evaluation of the content of any academic discipline or profession is
essential to ensure that knowledge and practice is based on fact. In recent
years I have come to question a construct that has been accepted by a wide
range of academic, research and professional entities: that the increasing
free proton concentration within contracting skeletal muscle is caused by the
increased production of “lactic acid”. One only has to read any of the
textbooks in exercise physiology or pure biochemistry to be informed that
when “pyruvic acid” is converted to “lactic acid”, the pK
of “lactic acid” results in an immediate, near complete dissociation of the
proton from the carboxylic acid functional group. This interpretation results in the logical
belief that the net result in vivo is the production of lactate ions and the
release of a proton. A generic
chemical equation used to support this explanation is as follows: Pyruvic
acid + NADH + H+ « lactic acid + NAD+ ® lactate-Na+ + NAD+ + H+ This
equation is typically extended to illustrate the bicarbonate buffering of the
proton from lactate, resulting in the non-metabolic production of carbon
dioxide (Brooks et al., 2000). Lactate-H
+ Na+ ® Na+-Lactate- + H+ H+ + HCO3- « H2CO3 « H2O + CO2 Physiology
is then extended to provide a cause-effect association between lactate
production, the development of acidosis, the added free H+ and CO2 stimulation of
ventilation, and the temporal alignment of the lactate and ventilatory
thresholds. The above
physiological and biochemical interpretations of a lactate-dependent acidosis
during exercise are so engrained that hundreds of papers published every year
directly or indirectly refer to it.
The error of the “lactic acidosis” construct in biochemistry and
physiology is that it is not based on fact.
Acidosis arises elsewhere than the lactate dehydrogenase (LDH)
reaction. The Biochemistry of Metabolic Acidosis Before I
commence my biochemical explanation of the development of acidosis during
exercise, I must stress that the concepts and explanations are not new. Credit is due to Gevers
(1977) for his initial publication and response (Gevers,
1979) to criticisms (Wilkie, 1979) of his alternate
views and explanations of metabolic acidosis in cardiac muscle. Subsequent reviews and commentaries of the
biochemistry of metabolic acidosis have substantiated the views of Gevers. For
example, Vaghy (1979) presented evidence for the
incorporation of cytosolic protons (hydrogen ions
free in the cytoplasm) into mitochondrial respiration within cardiac muscle,
and he theorized that any deficit in mitochondrial respiration would
contribute to acidosis. Dennis co-authored
a manuscript with Gevers 14 years later (Dennis et
al., 1991) that explained the importance of ATP hydrolysis to cytosolic proton production and accumulation. Similarly, additional researchers have
questioned the concept of a “lactic acidosis” and proposed a combination of
glycolysis and ATP hydrolysis to be the biochemical causes of proton release
and accumulation (Busa and Nuccitelli,
1984; Hochachka and Mommsen, 1983; Noakes, 1977; Zilva, 1978). It has
been almost 25 years since the original publication of Gevers
(1977), and there is no evidence in textbooks of the recognition that lactate
production does not cause acidosis. The “lactic acid” cause of acidosis, termed
a “lactic acidosis”, is still being taught in physiology and biochemistry courses
throughout the world. Researchers in
prestigious international journals are still using “lactic acid” and “lactic
acidosis” terminology (e.g., Hagberg, 1985; Juel, 1996, 1998; Katz and Sahlin,
1988; Stringer et al., 1994). Clearly,
a topic of this importance to basic and applied physiology, as well as to
clinical medicine, must be based on fact and not an unproven theory. A re-evaluation of the biochemistry of
exercise-induced metabolic acidosis is long overdue. Fundamentals of Acid-Base Physiology Prior to
explaining current and proposed interpretations of the biochemistry of
metabolic acidosis, I will clarify the difference between an acid and acid
salt. An acid is a molecule that at
neutral pH will release a proton into solution. Depending on the size of the molecule, the
proton comes from a specific type of chemical structure on the molecule,
typically called a functional group.
Larger acid molecules can have more than one acid functional group,
such as many of the amino acids. Some acid molecules are too small to contain
acid functional groups, but they are still acids (e.g., hydrochloric acid, HCl; perchloric acid, HClO4;
phosphoric acid, H3PO4). Figure 1 presents two examples of acid
functional groups within cellular metabolism: the carboxyl and phosphoryl groups.
The carboxyl group is theorized, within the “lactic acidosis”
construct, to play the greater role in cellular metabolic acidosis.
The
strength of an acid relates to the propensity for the molecule to release a
proton in solution, even when the solution is already acidic (pH below
7). Thus, strong acids will release a
proton until a relatively low pH is reached, at which time there is a dynamic
equilibrium between the protons that leave and re-attach to the acid
functional group of the molecule.
Consequently, to better understand the proton releasing potential of
an acid, it is necessary to know at what pH the release of the proton reaches
this dynamic equilibrium. This pH is
denoted as the negative log10 of the ionization constant,
abbreviated as pK'. At
equilibrium; HA « H+ + A-, where K
= products/substrates = ([H+] [A-]) / [HA] pK' = -log K = log(1/K) The pK, which represents the pH at which half of the acid
molecules are deprotonated (ionized), can be
determined in vitro by titration. As
you should be able to predict, strong acids or acid functional groups have a pK' much lower than 7, and weak acids have pK' values closer to 7.0.
The pK' values for a selection of acids and
acid functional groups are listed in Table 1.
After an
acid molecule loses a proton, it attains a negative ionic charge. To maintain charge neutrality, a cation ionically binds to the negative charge, resulting in an
acid salt. Due to the intracellular
and extracellular abundance of sodium (Na+) and potassium (K+), both being singly charged
cations, deprotonated acids are predominantly
sodium or potassium salts. Note that
in Table 1 the pK' of lactic acid is reported to be
3.86. Hence, the main form of “lactic
acid” in physiological systems is sodium lactate (La-Na+). Finally,
it should be emphasized that acid
production is not the only source of proton release within a cell. Protons can also be released from chemical
reactions, and I will show that this source of protons is the main cause of
acidosis in contracting skeletal muscle.
In addition, Stewart (1983) has clearly indicated that the movement of
charged ions across the muscle cell membrane can influence cell acid-base
balance, and this approach to understanding acid-base balance has been termed
the “strong ion difference”.
Additional research on the “strong ion difference” has shown that it
is associated with contributions to proton accumulation within contracting
muscle cells, presumably due to the efflux of potassium from muscle during
intense exercise (Lindinger and Heigenhauser,
1991). In this manuscript I focus on
proton release and consumption, and I will not consider further the influence
of the strong ion difference on pre-existing proton kinetics. The Source of Protons During Catabolism In Skeletal Muscle In the
sections that follow, I will explain the cytosolic
reactions of energy catabolism. I will
commence with the reactions of the phosphate energy system, and then the
reactions of glycolysis, finishing with the LDH reaction. For all reactions that involve either a
proton consumption or release, I provide structures to illustrate the
exchange of atoms, electrons and protons.
These atomically balanced equations are not provided in textbooks of
biochemistry or exercise physiology, which may explain why the biochemistry
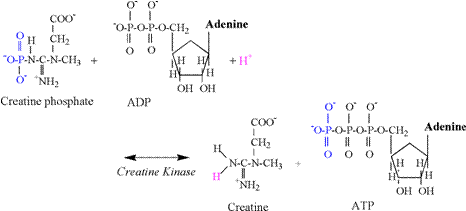
of acidosis is so poorly understood! Phosphagen Energy System: Creatine Kinase Reaction The
creatine kinase (CK) reaction is of vital importance to skeletal muscle
contraction. This reaction provides
the most immediate means to replenish ATP in the cytosol. Traditionally, the reaction has been
interpreted to be applicable mainly to the metabolic needs of intense
exercise, the transition to increased exercise intensities, or during
conditions of hypoxia. However,
creatine phosphate (CrP) probably assists in the
transfer of terminal phosphates throughout the cytosol,
as well as from the mitochondria to the cytosol.
This function is summarized as the linked reactions of the “creatine
phosphate shuttle” (Karlsson, 1971; Kent-Braun et
al., 1993). The chemical equation of
the CK reaction follows: Creatine
Phosphate + ADP + H+ « Creatine + ATP In vivo
the CK reaction is actually a coupled reaction involving breakdown of CrP and the phosphorylation of
ADP. It is incorrect to refer to this
in vivo reaction as hydrolysis of CrP. Hydrolysis of CrP
can occur in vitro, where water is required to provide the atoms and
electrons needed to produce creatine, inorganic phosphate (Pi), and a proton. The CK
reaction is referred to as an equilibrium reaction, as in vivo the free
energy change (DG) approximates zero. Thus, when the product of the molecules on
the left side of the equation increase relative to the right side of the
equation, such as during exercise of increasing intensity, the reaction
direction becomes exergonic in the direction of ATP
regeneration. The reaction reverses
direction during recovery from exercise. The
structural components of the creatine kinase reaction are detailed in Figure
2. The reaction involves the transfer
of a phosphate from CrP to ADP to form ATP. During exercise, increased rates of the CK
reaction actually cause a slight alkalinization of
skeletal muscle due to the consumption of a proton in the reaction (Karlsson, 1971, Dennis, 1991; Gevers,
1977). In order to reform the amine
terminal of creatine, a proton from solution is consumed in the reaction,
thus explaining the alkalinization. The carboxyl group of creatine (Cr) is
already ionized at physiological pH (Table 1) and does not contribute to the alkalinization.
The
biochemistry of the CK reaction indicates that 1 proton is consumed for every
phosphate transfer from CrP to ADP, forming
ATP. Thus, the CK reaction functions
as a small “sink” for protons, with an immediate capacity during exercise
equal to the number of CrP molecules that transfer
their phosphate to ADP. Phosphagen Energy System: Adenylate Kinase
Reaction For
increasing exercise intensities that extend into non-steady state conditions,
not only does the activity of the CK reaction increase, but the second
reaction of the phosphagen system also increases; the adenylate
kinase (AK) (or myokinase) reaction. The chemical equation of the AK reaction
follows: ADP
+ ADP « ATP + AMP The
production of AMP is important. AMP
increases the activity of phosphorylase, thereby
increasing glycogenolysis, as well as stimulating
increased activity of phosphofructokinase. The result of this stimulation is an
increased rate of glucose 6-phosphate formation to fuel glycolysis, and an
increased rate of glycolytic flux. As
will be discussed, this increased flux through glycolysis increases proton
release and eventually decreases cellular pH. Phosphagen Energy System: AMP Deaminase
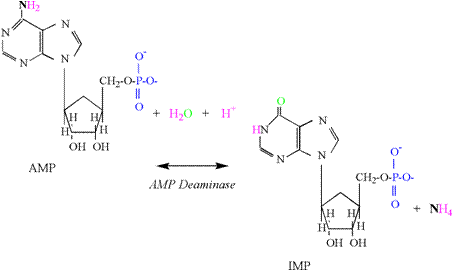
Reaction The
activity of the adenlyate kinase reaction is best
detected by increases in muscle adenosine monophosphate
(AMP) and inosine monophosphate
(IMP). The production of IMP results from an increased activity of the AMP deaminase reaction, which is activated by acidosis and
produces IMP and ammonia (NH4); AMP
+ H+ « IMP + NH4 The
reaction consumes a proton due to the initial formation of NH3
(Figure 3). The high pK of ammonia then results in addition of a proton. Adding the increased concentration of ADP
to the AMP and IMP produced in skeletal muscle (DADP + AMP + IMP) accounts for the
small decreases in ATP experienced during intense exercise to fatigue.
It is important to recognize that the AK and
AMP deaminase reactions reflect an inability for
mitochondrial respiration to totally replenish ATP within the cytosol of the cell.
Research indicates that these cellular conditions are associated with
the greatest ATP regeneration from the phosphagen system and glycolysis, and
coincide with a rapid increase in lactate and proton accumulation (decreased
pH) (Karlsson, 1971; Sahlin,
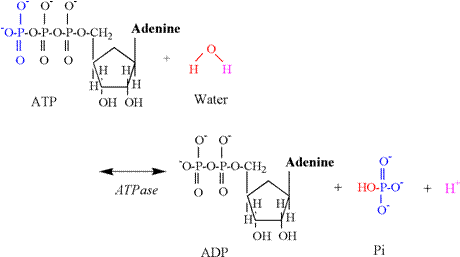
1978; Sahlin et al., 1987; Katz and Sahlin, 1988). Phosphagen Energy System: ATP Hydrolysis Muscle
contraction necessitates the breakdown (hydrolysis) of ATP to ADP and Pi (HPO4-2).
The enzyme for this reaction is myosin ATPase,
and the chemical equation follows: ATP
+ H2O « ADP + Pi + H+ The
proton release associated with this reaction results from the involvement of
water, which is necessary to provide an oxygen atom to bind to the terminal
phosphate of ADP and a hydroxyl group which binds to Pi (Figure 4). A proton is released in conditions of
physiological pH, as the pK's of the remaining
oxygen atoms of the phosphate group are too low to be protonated
(Table 1). ATP
hydrolysis during muscle contraction is the primary stimulus for increasing
energy catabolism. The primary function of energy catabolism
appears to be maintenance of the cellular ATP concentration. At the onset of moderate-intensity
exercise, the phosphagen system and glycolytic ATP regeneration maintain
cellular ATP until mitochondrial respiration is adequately stimulated. The
products of ATP hydrolysis can all be used by the cell under steady-state
conditions. The cytosolic
ADP is involved in the transfer of phosphate groups from mitochondrial ATP to
cytosolic Cr, and to reform ATP as described in the
section on the CK reaction. ADP is
also directly transported into the mitochondria as a substrate for oxidative phosphorylation.
The Pi is used as a substrate for glycogenolysis
(phosphorylase reaction) and the glyceraldehyde 3-phosphate dehydrogenase reaction of
glycolysis. In addition, the Pi can
also be transported into the mitochondria, where it is needed as a substrate
for oxidative phosphorylation. The protons from ATP hydrolysis can also be
shuttled into the mitochondria via the malate-aspartate
or glycero-phosphate shuttles, or by direct
transport via proton transporters (e.g., the monocarboxylate
lactate-proton transporter). The
protons then assist in the maintenance of the proton gradient between the
mitochondrial inner-membranous space and matrix.
The free
inorganic phosphate is not a strong acid, because all but one proton has
dissociated at physiological pH, leaving HPO4-2.
Interestingly, inorganic phosphate can function as a buffer as pH
falls, because the pK' of one of the hydroxyl
functional groups is 6.82 (Table 1).
The pH-dependent buffering potential of Pi is revealed in
31-Phosphorous magnetic resonance spectroscopy (31P-MRS), via a shift in the frequency spectrum of Pi
when the Pi becomes protonated. This shift is used to calculate cytosolic pH using a modified Henderson-Hasselbalch equation (Kent-Braun et al., 1993). Phosphagen Energy System: Summary During
exercise of increasing intensity into non-steady state, the activity of the
CK reaction increases. The CK reaction
decreases CrP, at the same time consuming a
proton. Together with the AK reaction,
cellular ATP concentrations are well maintained, despite the inadequacy in the rate of ATP regeneration by
mitochondrial respiration. These
cellular conditions are also associated with an increase in Pi. However, the accumulation of this molecule
is not a result of the CK reaction as is generally believed within sports and
exercise science, but results from a net dephosphorylation
of ATP during muscle contraction. An
increasing cellular Pi concentration therefore indicates that the cell is
lagging behind in the regeneration of ATP from mitochondrial respiration, as
Pi is not re-used by glycolysis or transported into the mitochondria as a
substrate for oxidative phosphorylation. When the
cell develops an inability to supply all cellular ATP needs from
mitochondrial respiration, it is a gradual process and not readily detected
by assaying ATP due to the effectiveness of the CK and AK reactions, as well
as an increasing rate of ATP regeneration from glycolysis. Nevertheless, ATP hydrolysis releases a
proton, and when unmatched by an equal rate of mitochondrial respiration
derived ATP regeneration, this proton is left to accumulate in the cytosol (Kent-Braun et al., 1993). The power of proton release from ATP
hydrolysis is in direct proportion to the rate of ATP turnover. However, to a small extent the proton yield
from ATP hydrolysis is reduced by the proton consumption of the CK and AMP deaminase reactions.
As acidosis increases (pH<6.9), added proton buffering is provided
by phosphate groups (Pi, hexose and triose phosphates).
Glycolysis The
reactions of glycolysis are listed in Table 2. Two reactions of Phase 1 and one reaction
of Phase 2 release protons, whereas one reaction of Phase 2 consumes
protons. Consequently, when starting
from glucose or glycogen, glycolysis yields a net of two protons per glucose
flux to 2 pyruvate. Increasing glycolytic flux increases net proton release
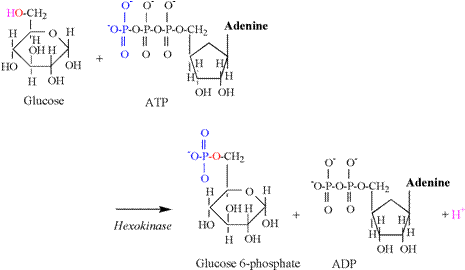
and the need for proton buffering. The hexokinase reaction is the first proton releasing
reaction of glycolysis, and is illustrated in Figure 5. The hydroxyl group of the sixth carbon is
split during this reaction, releasing a proton. The oxygen and electron remain to accept
the phosphate group transferred from ATP.
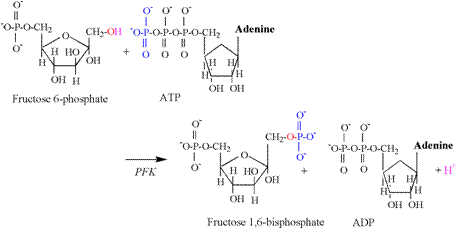
The second proton releasing reaction of
glycolysis is catalyzed by phosphofructokinase
(PFK), and is illustrated in Figure 6.
As for the hexokinase reaction, the hydroxyl
group of the first carbon is split, releasing a proton, followed by the
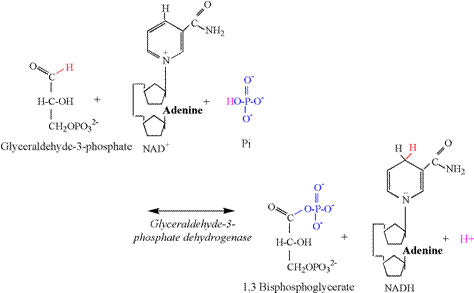
acceptance of the phosphate group transferred from ATP. Glyceraldehyde 3-phosphate dehydrogenase (G3PDH) catalyzes the third
proton releasing reaction of glycolysis (Figure 7). The aldehyde
group of the third carbon is oxidized by NAD+, resulting in the removal of two
electrons and a proton. A proton is also removed from Pi, allowing the Pi to
bind to the third carbon, forming 1,3 bisphosphoglycerate. The G3PDH
reaction is functionally and bioenergetically
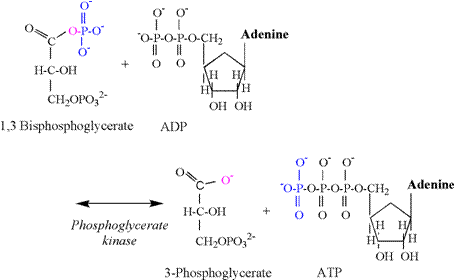
coupled to the phosphoglyerate kinase (PGK)
reaction (Figure 8). Note that the PGK
reaction produces the first acid intermediate of glycolysis;
3-phosphoglycerate. In an early
edition of his textbook, Lehninger (1993) explained
that this reaction produced a proton via ionization of 3-phosphoglycerate at
physiological pH. However, this
presentation should not be applied to in-vivo conditions, as Lehninger was illustrating the reaction mechanism of
hydrolysis, which in vitro occurs without the coupling of the reaction to ADP
phosphorylation.
The PGK reaction involves a simple phosphate
transfer from the first carbon of 1,3 bisphosphoglycerate
to ADP, forming ATP. An oxygen and
electron remain on the carboxylic acid functional group of 1,3 bisphosphoglycerate.
There is no proton involved in this transfer, and 3-phosphoglycerate
is formed devoid of a proton. This
same carboxyl group remains unprotonated for the
remaining intermediates of glycolysis.
This important biochemical fact means that there never was a proton
released by the carboxyl group of 3-phosphoglycerate or any of the following
glycolytic intermediates. Thus there
is no proton associated with the carboxyl group when lactate is produced. The only conclusion to be made from this
biochemical fact is that it is
impossible for lactate production, or that of any “downstream” carboxylic
intermediate from 3-phosphglycerate, to cause the release of a proton and a
subsequent acidosis. This fact
alone obliterates the notion of a “lactic acidosis”.
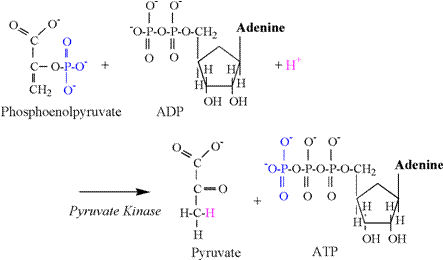
The pyruvate kinase reaction consumes a
proton, and is illustrated in Figure 9.
The phosphate group attached to the second carbon of phosphoenolpyruvate is transferred to ADP, forming
ATP. The preferred chemical state of
pyruvate is an enol form (second carbon double bond
to oxygen), and a proton is required from solution to bind to the third
carbon to complete the chemical structure.
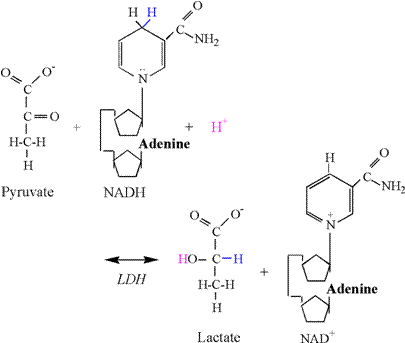
The Lactate Dehydrogenase Reaction Once
pyruvate is produced, it can be transported into the mitochondria and
oxidized via the pyruvate dehydrogenase complex reaction, or reduced to
lactate in the cytosol via LDH. The chemical equation of the LDH reaction
follows: Pyruvate
+ NADH + H+ « lactate + NAD+ The
reduction of pyruvate involves the addition of 2 electrons and 1 proton from
NADH, and 1 proton from the cytosol (Figure
10). The second carbon of pyruvate is
reduced by the addition an electron and proton from NADH, forming the
covalent bond to hydrogen. Another
electron from NADH and a proton from solution are used to form the hydroxyl
group. As previously explained, there
is no proton associated with the carboxyl group, and therefore no proton
release and ionization. Thus, the LDH
reaction consumes a proton, thereby functioning as a sink for protons
produced in catabolism and ATP hydrolysis.
The Balance of Proton Production and Consumption in Muscle Contraction Based on
the metabolic biochemistry presented, an estimation of the balance of proton
production and consumption (including buffering) can be made. However, such estimates only represent the
proton exchange resulting from the reactions of the phosphagen and glycolytic
systems, as modified by carbohydrate oxidation in mitochondrial
respiration. Added proton exchange
occurs during amino acid oxidation and associated amination
and deamination reactions. In addition, blood and tissue acid-base
balance is further complicated during ketosis. Nevertheless, during short-term intense
exercise to fatigue, it is fair to conclude that the phosphagen and
glycolytic systems represent the bulk of proton exchange. When
accounting for proton production in the cytosol during
muscle contraction, the sources are glycolysis and ATP hydrolysis. For proton consumption, the contributors
are the CK reaction, AMP deaminase reaction,
mitochondrial respiration, Pi and additional intracellular buffers, and
proton efflux from the cell (Table 3).
It should be noted that as pH decreases, Pi (free and hexose- and triose phosphates)
becomes a stronger buffer of protons due to the increased proportion of the
molecules that consume a proton from solution (forming H2PO3-).
For simplicity, I have not based calculations on fractional
contributions to proton exchange. This
decision is based on past research and reviews on this topic which reveals
that magnesium is bound to all adenylates, thereby
lowering pK values to non-physiologically acidic
levels (Karlsson, 1971). Furthermore, the proton buffering of Pi is
minimal, and fractional representation of this component causes minimal
change to the overall tally of proton exchange.
The data
of Table 3 refer to intense exercise to fatigue, and are derived from the
research of Spriet et al. (1987, 1987) and Medbo et al. (1993).
I have used the data of Medbo et al. based
on 3 min of cycle ergometry at 120% VO2max to calculate a balance
of proton release and consumption. As
the glycolytic contribution to ATP turnover was not calculated by Medbo, I estimated it based on a 60% glycolytic
contribution to total ATP turnover as recommended by Spriet
(1990). When the
sum of all proton releasing components and proton consuming components are
tallied, ~145 mmol H+/kg/3 min remains for handling by buffers and extracellular transport
from the cell. This seems appropriate,
as research has produced values for muscle buffering between 40 to 80 mmol H+/L/pH. As this is a capacity that adapts with
training, using a high value of 80 mmol H+/L/pH is reasonable, which
approximates to 59 mmol H+/kg/pH. It is difficult to
convert this to a capacity, but with a drop in muscle pH from 7 to 6.4, this
amounts to 35.4 mmol H+/kg. Consequently, proton
efflux from muscle must approximate 110 mmol H+/kg/3 min, or 37 mmol H+/kg/min; a value that is distributed
among passive proton removal, bicarbonate buffering, and the proton
transporters (Na+, HCO3-, lactate). Unfortunately,
research of the proton efflux from human muscle of a heterogenous
fiber type is not extensive, and it is difficult, if not impossible, to gauge
the validity of the 37 mmol H+/kg/min estimate of proton efflux (Brooks,
2000; Juel, 1996, 1998). As the
free protons remaining in solution within a cell do not amount to a large
concentration (e.g., a pH decrease from 7.0 to 6.4 results in a proton
accumulation of 0.3 mmol/L, or approximately 0.22 mmol/kg), the value of 30 to 40 mmol H+/kg/min for proton efflux is
high. Nevertheless, the aforementioned
calculations of proton balance are more realistic than if acidosis was
dependent on lactate production. Assuming
that 25% of total muscle lactate is removed during 3 min of intense exercise
to fatigue, then approximately 40 mmol/kg lactate/3 min is produced. When accounting for the added proton
consuming reactions of metabolism (i.e., ignoring that the LDH consumes a
proton!), net proton release from lactate during 3 min of exhausting exercise
would only amount to 17 mmol/kg (40–20–3 = 17 mmol/kg). Based on the muscle buffer capacity (35.4
mmol H+/kg),
and the above estimate of proton efflux from muscle (110 mmol H+/kg/3 min), this value for
lactate-related proton release is only 15% of the total protons accounted for
by buffering and efflux. Clearly, the
concept of a “lactic acidosis” is not supported by biochemistry, or from data
of muscle lactate production, and proton buffering and efflux during intense
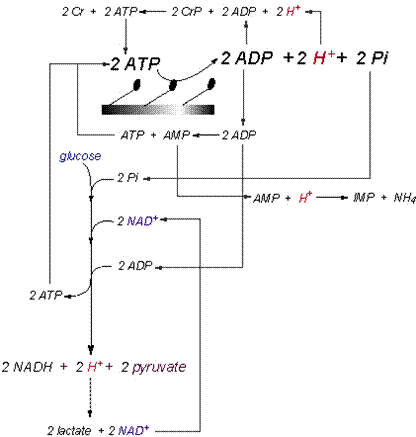
exercise to fatigue. Summary of Cytosolic Proton Exchange The balance of the proton-releasing and
proton-consuming reactions of catabolism in skeletal muscle needs to be
presented in a manner that expresses the simultaneous activity of all
pertinent reactions. I have tried to
do this for the cytosolic reactions in Figure 11,
and this illustrative summary of the prior content of this manuscript needs
to be applied to the capacities and power of proton release and consumption
summarized in Table 3.
During
exercise of low intensity, glycolytic flux is low, the predominant substrate
for energy catabolism is fatty acids, and consequently, a muscle’s ATP needs
are met largely by mitochondrial respiration.
With an increase in exercise intensity, blood flow and glucose uptake
into skeletal muscle increases. In
addition, free calcium and Pi increase slightly, thereby increasing the
activity of phosphorylase. The additional glucose uptake and
increasing rate of glycogenolysis increase
glycolytic flux, and in so doing, decrease the relative contribution of fatty
acid oxidation to total ATP regeneration.
With an increasing glycoytic flux, yet still
a steady state intensity, there is also an increase in proton release.
However, the protons are consumed by lactate production and transport into
the mitochondria for use in oxidative phosphorylation. As the
exercise intensity increases to now exceed the threshold point for the
handling of the cytosolic ATP demand by
mitochondrial respiration, there are transient increases in ADP, causing an
increased rate of the CK reaction. In
addition, Pi begins to accumulate, providing added substrate for glycogenolysis and glycolysis, further increasing
substrate flux through glycolysis.
These events lead to rapid increases in proton release due to an
increasing dependence on glycolysis for sustaining the cellular ATP
concentration. Consequently, the main
cause of an increasing proton release is the greater rate of glycolytic flux,
plus the now increasing dependence on glycolytic ATP turnover. The increasing substrate flux through
glycolysis, accompanied by decreases in the cytosolic
redox (NAD+/NADH) results in an increased
rate of lactate production (Sahlin et al., 1987).
Lactate
production is beneficial for regenerating NAD+ as well as consuming a
proton. Nevertheless, the capacity of
the LDH reaction to maintain cytosolic redox and retard a worsening acidosis depends on the
maximal rate of proton efflux from the cell.
Fortunately, the lactate transporter also co-transports a proton. Additional proton transporters also exist
(Na+ and
HCO3-). Thus, lactate production has a third advantage: assisting proton efflux
from muscle. Despite these
benefits, the lactate-proton transport is rate limiting, and as lactate
accumulates in the cytosol, the bioenergetics of
the LDH reaction become less favorable, and the rate of lactate production
decreases. During sustained intense
exercise, the rate of pyruvate and lactate production is also decreased due
to a reduction in the rates of glycogenolysis and
glycolysis, which occur as early as 30 s into a 3-min bout of intense
exercise. The accummulation
of pyruvate in the cytosol of the cell, and the
accumulation of acetyl units in the mitochondria reflect a glycolytic
activity that does not end in lactate production or the complete oxidation of
glucose carbons. Thus, added protons
accumulate and acidosis is worsened. Application of Biochemistry of Acidosis to Exercise Physiology Clearly, there is no biochemical evidence for
lactate production releasing a proton and causing acidosis. Nor is there any evidence that lactate
production increases in equal amounts to the number of protons released
within and from skeletal muscle.
Consequently, the cause of acidosis should be taught to be a result of
exercise intensities that are now non-steady state. Such conditions result in further increases in the rate of glycolysis, and
increased dependence on cytosolic ATP turnover due
to a mismatch between the rate of ATP demand (muscle contraction) and supply
from mitochondrial respiration.
These
cellular conditions have large implications to how we understand exercise
physiology. For example, lactate production retards, not worsens
acidosis. A greater capacity to
produce and remove lactate from the cell would delay the onset of acidosis. This means that during intense exercise,
high lactate production is beneficial to the athlete, especially when
accompanied by a high capacity for lactate and proton transport from the
cell, capacities that are known to increase with both endurance and
power/sprint training (Juel, 1998). The
temporal alignment between the cellular conditions leading to acidosis and an
increased production of lactate are not changed by this biochemical
explanation of acidosis. Lactate is
obviously a good indirect marker of an alteration in cellular metabolism
causing acidosis and a non-steady state cellular metabolic milieu. However, lactate production does not cause
the acidosis. Another
application of the biochemistry of metabolic acidosis relates to the
recruitment of fast twitch motor units.
As exercise intensity increases, Type IIa
and IIb motor units become progressively
recruited. As the muscle fibers of
these motor units have a lower mitochondrial density than Type I fibers, they
are more reliant on glycolysis and cytosolic ATP
turnover. As these two traits combine
to increase the net rate of proton release from catabolism, a considerable
proton production ensues during the exercise intensities associated with
increased Type II motor unit recruitment.
As such, type II fibers contribute to acidosis, not because they
produce more lactate, but because they have less mitochondria to assist in
ATP regeneration, and uptake of protons. The
biochemistry of acidosis also has clinical implications. Obviously, attempting to prevent acidosis
by inhibiting lactate production will worsen, not prevent, acidosis. The best means to prevent or delay acidosis
is to decrease reliance on glycolysis, improve the contribution of ATP
turnover from mitochondrial respiration, and increase the capacity of proton
buffering and lactate-proton removal.
The former strategies are typical for endurance training, and the
latter strategy is applicable to strength and power training. However, clinical strategies would relate
to increasing lipid oxidation by raising blood free fatty acids, or
stimulating mitochondrial function. Why is Lactic Acid Still Thought to Cause Acidosis? Despite the biochemical realities I have presented, the fact remains
that most academics and researchers within the pure and applied fields of
physiology and biochemistry still think that lactate production causes
acidosis. One major explanation for this fact is that the biochemistry sections
in textbooks do not present chemical equations balanced for protons and
water. Thus, I had to apply my own
knowledge of organic chemistry to derive the diagrams of chemical reactions
presented in this article. Most PhDs
and physicians simply have not been educated correctly about the biochemistry
of energy metabolism in skeletal muscle.
Ironically, even the main textbooks of biochemistry do not devote a
chapter to explaining the biochemistry of metabolic acidosis. Specific coverage is warranted for this important
topic. Textbooks of exercise physiology are even worse in their treatment of
the biochemistry of acidosis. Acidosis
is attributed to the production of “lactic acid” which, when ionized at
cellular pH, releases a proton into solution.
This explanation is made without any support from research or
biochemistry. I have clearly shown the shortcomings of this explanation. Until
textbooks detail the realities of the biochemistry of acidosis, the myth of
lactic acidosis will continue. Rob’s
Figure 11 is an elegant summary of the flow of compounds involved in energy
consumption and anaerobic energy production during exercise. If we follow the
fate of a molecule of glucose down this pathway, we end up with two
lactates. Along the way, we use up two
ADP and generate two ATP, and that’s all.
There is no nett production of H+.
But ATP and ADP don’t change, because muscle contraction breaks the
two ATP back down to two ADP, two Pi, and, of course, two H+.
So we end up with two lactates and two H+ for each molecule of
glucose. We’ve turned glucose into
lactic acid, but the lactate and the acid come from different places: the H+ from breakdown of ATP, and the
lactate from breakdown of glucose. That appears to be Rob’s view, and it
seems perfectly reasonable. But it's also perfectly reasonable to argue that
the H+ comes
from glucose, one way or another. The
H+ produced in the hydrolysis of ATP
comes from water, sure, but matter is conserved. Somewhere somehow in the breakdown of
glucose an H+ is
transferred to water. Ultimately, a
molecule of glucose ends up as two molecules of lactate and two H+.
I have no real objection to “lactic acidosis”. Will first contacted me about this topic after he read one of my
abstracts from the 2001 annual meeting of the American College of Sports
Medicine. This article arose from our
interaction. In my view, and in the view of many exercise physiologists and
biochemists, “lactic acid” does not directly cause exercise-induced metabolic
acidosis. Will and I have been
grappling with the reasoning behind this view, and whether it has any real
meaning to how we, as exercise and sports science professionals, interpret
exercise-induced lactate production and acidosis. Will’s comment is consistent with the
biochemical evidence. Certainly, when looking at metabolism from a general perspective,
intense exercise induces acidosis that coincides with an accumulation of
lactate. My problem with the concept of a “lactic acidosis” is that it is yet
another example of an exercise and sports science oversimplification of
biochemical fact for the sake of simplicity and expedience. It bothers me that Will still wants to
generalize facts to an association between acidosis and lactate. In my experience, the generalizations
within exercise and sports science are negative reflections on us, and it is
no wonder that our field is viewed poorly by many (not all) academics and
researchers in the pure sciences.
Exercise-induced metabolic acidosis is far more complex than to lay
blame on one reaction and product, and we should accept the challenge of
being true scientists to explain the reality of cellular acidosis. The point is not whether it really makes a major difference, or
whether the net result is or is not lactate accompanied by protons. Let’s teach the facts and then let us see
where the truth takes us: in education, in research, and in clinical
applications. Finally, it is important to realize that although Will’s assessment of
the lactate and proton balance is reasonable (2 lactate + 2 protons), the
fact remains that because the protons do not come from the production of
lactate, there is potential for an uncoupling of the ratio of protons to
lactate. For example, lactate
production will underestimate the net proton release when the pyruvate that
does not enter the mitochondria is incompletely converted to lactate
(increase in cellular pyruvate). Similarly, when pyruvate is transported into
the mitochondria, converted to acetyl CoA, and
these acetyl groups accumulate in the mitochondria due to an insufficiency of
mitochondrial respiration, added protons also accumulate in the cytosol due to the absence of lactate production from
these carbons and the accompanied proton consumption. Extra proton
accumulation also occurs in the cytosol from the
hydrolysis of ATP gained from the adenylate kinase
reaction. For all these reasons,
proton release is greater than lactate production. |
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
References Brooks GA
(2000). Intra- and extra-cellular lactate shuttles. Medicine and Science in
Sports and Exercise 32, 790-799 Busa
WB, Nuccitelli R (1984). Metabolic regulation via intracellular
pH. American Journal of Physiology
246, R409-R438 Dennis
SC, Gevers W, Opie LH
(1991). Protons in ischemia: Where do
they come from; where do they go to?
Journal of Molecular and Cell Cardiology 23, 1077-1086 Gevers
W (1977). Generation of protons by
metabolic processes in heart cells. Journal
of Molecular and Cell Cardiology 9, 867-874 Gevers
W (1979). Generation of protons by
metabolic processes other than glycolysis in muscle cells: a critical view
[letter to the editor]. Journal of
Molecular and Cell Cardiology 11, 328 Hagberg
H (1985). Intracellular pH during
ischemia in skeletal muscle: relationship to membrane potential,
extracellular pH, tissue lactic acid and ATP.
Pflugers Archives 404, 342-347 Hochachka
PW, Mommsen TP (1983). Protons and anaerobiosis. Science 219, 1391-1397 Juel C
(1996). Lactate/proton co-transport in skeletal muscle: regulation and
importance for pH homeostasis. Acta Physiologica Scandinavica 156, 369-374 Juel C
(1998). Muscle pH regulation: role of training. Acta
Physiologica Scandinavica. 162, 359-66 Karlsson
J (1971). Lactate and phosphagen concentrations in working muscle of man. Acta Physiologica Scandinavica Supplementum
358, 1-72 Katz A, Sahlin K (1988). Regulation of lactic acid production
during exercise. Journal of Applied Physiology 65, 509-18 Kent-Braun
JA, Miller RG, Weiner MW (1993). Phases of metabolism during progressive
exercise to fatigue in human skeletal muscle. Journal of Applied Physiology
75, 573-80 Lehninger
AB, Nelson DL, Cox MM (1993). Principles of biochemistry (second edition).
New York, NY: Worth Publishers Lindinger
MI, Heigenhauser GJ (1991). The roles of ion fluxes
in skeletal muscle fatigue. Canadian Journal of Physiology and Pharmacology
69, 246-253 Medbo
JI, Tabata I (1993). Anaerobic energy release in
working muscle during 30 s to 3 min of exhausting bicycling. Journal of
Applied Physiology 75, 1654-1660 Nelson
DL, Cox MM (2000). Lehninger principles of
biochemistry (third edition). New York, NY: Worth Publishers Noakes TD
(1997). Challenging beliefs: ex Africa semper
aliquid novi. Medicine and Science in Sports and Exercise
29, 571-590 Sahlin
K (1978). Intracellular pH and energy
metabolism in skeletal muscle of man. Acta Physiologica Scandinavica 455, S7-50 Sahlin
K, Katz A, Henriksson J (1987). Redox state and
lactate accumulation in human skeletal muscle during dynamic exercise. Biochemical Journal 245, 551-556 Spriet
LL, Sunderland K, Bergstrom M, Hultman E (1987).
Anaerobic energy release in skeletal muscle during electrical stimulation in
men. Journal of Applied Physiology 62, 611-615. Spriet
LL, Sunderland K, Bergstrom M, Hultman E (1987).
Skeletal muscle glycogenolysis, glycolysis, and pH
during electrical stimulation in men. Journal of Applied Physiology 62,
616-621 Spriet
LL (1990). Anaerobic meatabolism in human skeletal
muscle during short-term, intense activity. Canadian Journal of Physiology
and Pharmacology 70, 157-165 Stewart
PA (1983). Modern quantitative acid-base chemistry. Canadian Journal of
Physiology and Pharmacology 61, 1444-1461 Stringer
W, Wasserman K, Casaburi R, Porszasz
J, Maehara K, French W (1994). Lactic acidosis as a
facilitator of oxyhemoglobin dissociation during
exercise. Journal of Applied Physiology 76, 1462-1467 Stryer
L (1988). Biochemistry (third edition). New York, NY: W.H. Freeman Vaghy
PL (1979). Role of mitochondrial
oxidative phosphorylation in the maintenance of
intracellular pH. Journal of Molecular
and Cell Cardiology 11, 933-940 Wilkie
DR (1979). Generation of protons by
metabolic processes other than glycolysis in muscle cells: a critical
view. Journal of Molecular and Cell
Cardiology 11, 325-330 Zilva
JF (1978). The origin of the acidosis
in hyperlactataemia. Annals of Clinical Biochemistry 15, 40-43
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||